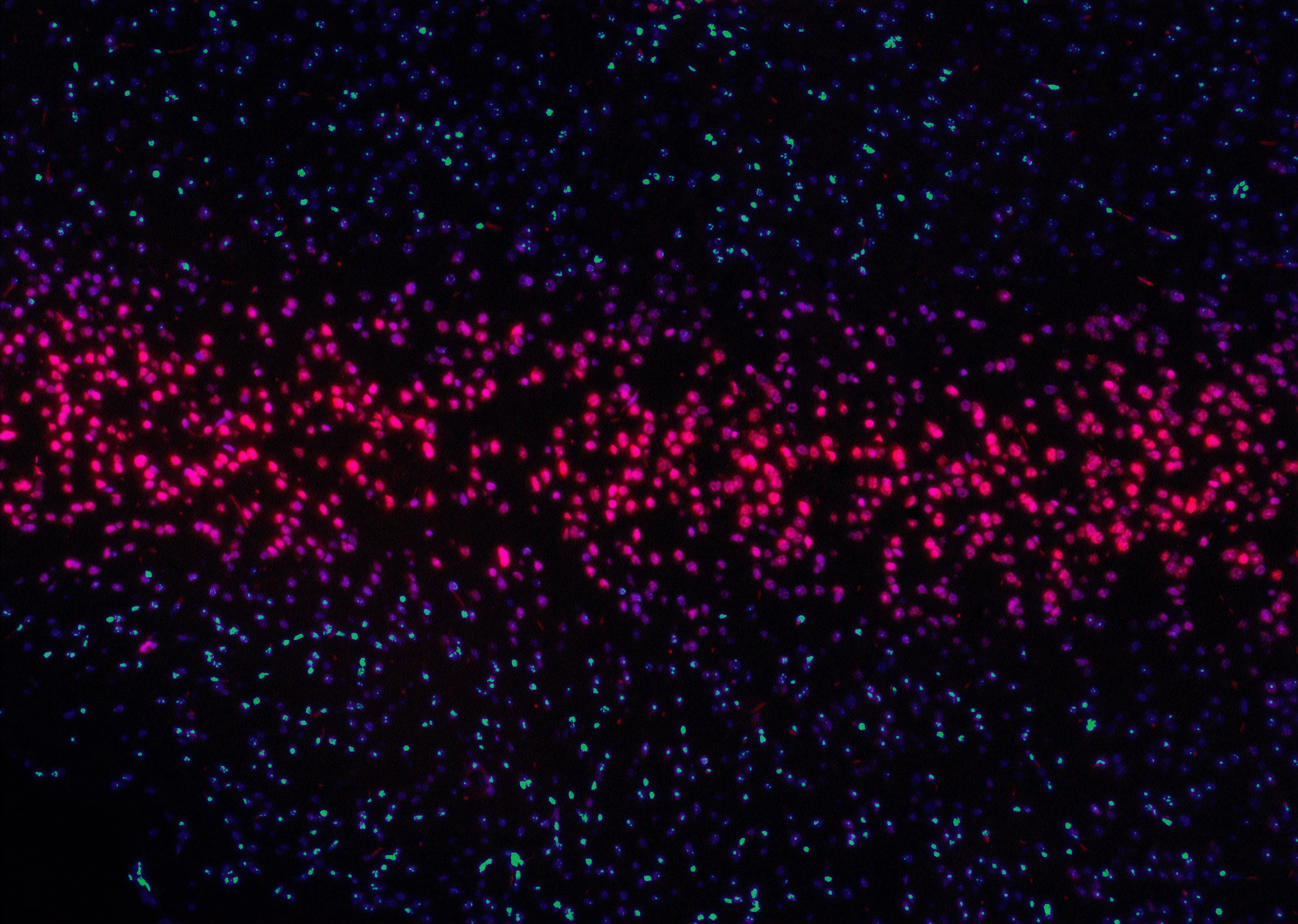
In a study exploring the interaction between genetics and Alzheimer’s disease (AD) pathology, researchers discovered how the APOE3 Christchurch (APOE3ch) mutation protects against AD. Through support from Cure Alzheimer’s Fund, an APOE3ch mouse model was created, allowing researchers to investigate the influence of the Christchurch mutation on amyloid and tau pathology. The findings showcase how APOE3ch revs up the efficiency of microglia surrounding amyloid plaques to remove aggregated tau. Although APOE3ch reduced the levels of amyloid beta, its real power was the ability to prevent the spread of tau, which precedes neuronal death and dementia. These findings reinforce the potential of APOE3ch as a genetic shield against AD, and present a novel perspective on the genetic mechanisms leading to AD resilience.
In 2019, a Nature Medicine article described the intriguing case of a woman who, despite inheriting a gene mutation responsible for a rare, early-onset form of AD, escaped developing AD dementia. In fact, she remained cognitively normal into her 70s and only then developed mild cognitive impairment, which lasted until her death from cancer at age 78. Postmortem analysis of her brain showed unusually high levels of amyloid beta but limited levels of tau and neurodegeneration. The key to her resilience appeared to rest with the fact she also had a specific mutation in both copies of the APOE3 gene. This mutation is known as the APOE3 Christchurch (APOE3ch) mutation.
However, because this unique combination of mutations only was found in one person, scientists could not prove that APOE3ch prevented the development of AD dementia. Now, with the development of an APOE3ch mouse model, researchers have been able to study the influence of APOE3ch on amyloid and tau pathology and provide evidence of how APOE3ch safeguards against dementia.
The woman highlighted in the Nature Medicine article was part of a large, extended Colombian family in South America with a high prevalence of early-onset AD due to a mutation in the Presenilin I (PSEN1) gene. This mutation causes the overproduction of amyloid beta. Members of this kindred who inherit the PSEN1 mutation develop symptoms of AD when they enter their 40s, much earlier than the more common late-onset form of AD, which typically affects people in their mid-60s and beyond. Although AD can be characterized as early- or late-onset depending on when clinical symptoms appear, they all share the same classic pathological hallmarks of amyloid plaques and neurofibrillary (tau) tangles.
A significant genetic factor for late-onset AD is the APOE gene, which has three forms:
- APOE2, known to be protective.
- APOE4, which increases risk.
- APOE3, which typically does not influence risk.
In the case of the Colombian woman, a mutation in APOE3 appears to confer remarkable resilience against AD. To study how this was possible, researchers—with support from a grant provided by Cure Alzheimer’s Fund—created a mouse model containing two copies of APOE3ch and modified it to overproduce amyloid beta. Since mice don’t naturally replicate the tau pathology seen in humans, a small amount of human tau was introduced into the brains of the mice which, in the presence of amyloid beta, would pathologically spread through the brain.
However, this is not what researchers saw in the APOE3ch model. Like the Colombian woman, these mice had extensive amyloid beta throughout their brains but very little tau. The Christchurch mutation energized the brain’s natural immune cells, the microglia, to engulf and degrade tau more efficiently. They were so efficient that tau was prevented from spreading.
This discovery is important because it showcased how APOE3ch blocked the transition between amyloid beta buildup and tau spread. In AD, amyloid beta accumulates in the brain for decades before symptoms appear. The authors of this study, along with many other scientists in the Alzheimer’s disease research community, theorize that the buildup of amyloid beta eventually triggers tau to tangle and spread throughout the brain. As a result, the brain becomes inflamed, neurons die, and memory and cognition suffer. The question of why amyloid beta triggers tauopathy is not clearly understood. However, the spread of tau is a greater indicator of cognitive impairment than the accumulation of amyloid beta. By impacting the activity of microglia, APOE3ch blocked the destructive cascade that leads to AD dementia.
While APOE3ch modestly decreased the amount of amyloid beta present compared with mice that didn’t have the Christchurch mutation, its effect on microglia holds the most promise for disease prevention. Developing a therapy to mimic APOE3ch’s impact on microglia efficiency could harness its power to render amyloid beta accumulation harmless and prevent AD-associated dementia.
Published in Cell:
APOE3ch Alters Microglial Response and Suppresses Ab-Induced Tau Seeding and Spread
Marco Colonna, M.D., Washington University in St. Louis
Jason D. Ulrich, Ph.D., Washington University in St. Louis
David Holtzman, M.D., Washington University in St. Louis