Despite high levels of amyloid beta plaques and tau tangles in their brain, some individuals never develop symptoms of dementia in their lifetime. Resilience to pathology leading to Alzheimer’s disease (AD) may be associated with reduced levels of neuroinflammation, thought to be universal in symptomatic AD. But what in the AD brain could be triggering excessive neuroinflammation has remained elusive. A breakthrough study uncovers the origin of resilience to the pathology leading to AD.
***************
Resilient brains provide a rare glimpse into the protective mechanisms that may be at play in individuals who would be expected to have dementia as a result of the heavy burden of plaques and tangles. The pathology that leads to Alzheimer’s disease begins many years prior to first symptoms. Amyloid beta spreads throughout the brain as bundles of plaques, followed by the formation of tau tangles, resulting in synapse loss. Synapses are the points of communication between neurons and are considered the fundamental information-processing units of the brain’s wiring that underlie memory and cognition. Significant loss of synapses—neurodegeneration—is the immediate cause of cognitive decline.
In postmortem evaluation, some brains have significant levels of amyloid plaques and tau tangles while remaining asymptomatic. The research suggests that resilience to AD pathology stems from reduced levels of neuroinflammation that preserve synapses and stave off neurodegeneration.
Neuroinflammation is mediated by the brain’s glial cells, the microglia and astrocytes. Under normal conditions, microglia act as sentries and clean the brain of debris and toxic molecules, while astrocytes nurture and support neurons. Under pathological conditions, microglia and astrocytes become inflammatory. When a distress signal is present, these cells migrate to the site and remove the threat. But in the case of excessive neuroinflammation, microglia and astrocytes become overactive and, in their response, exceedingly eliminate synapses—leading to neurodegeneration as seen in AD.
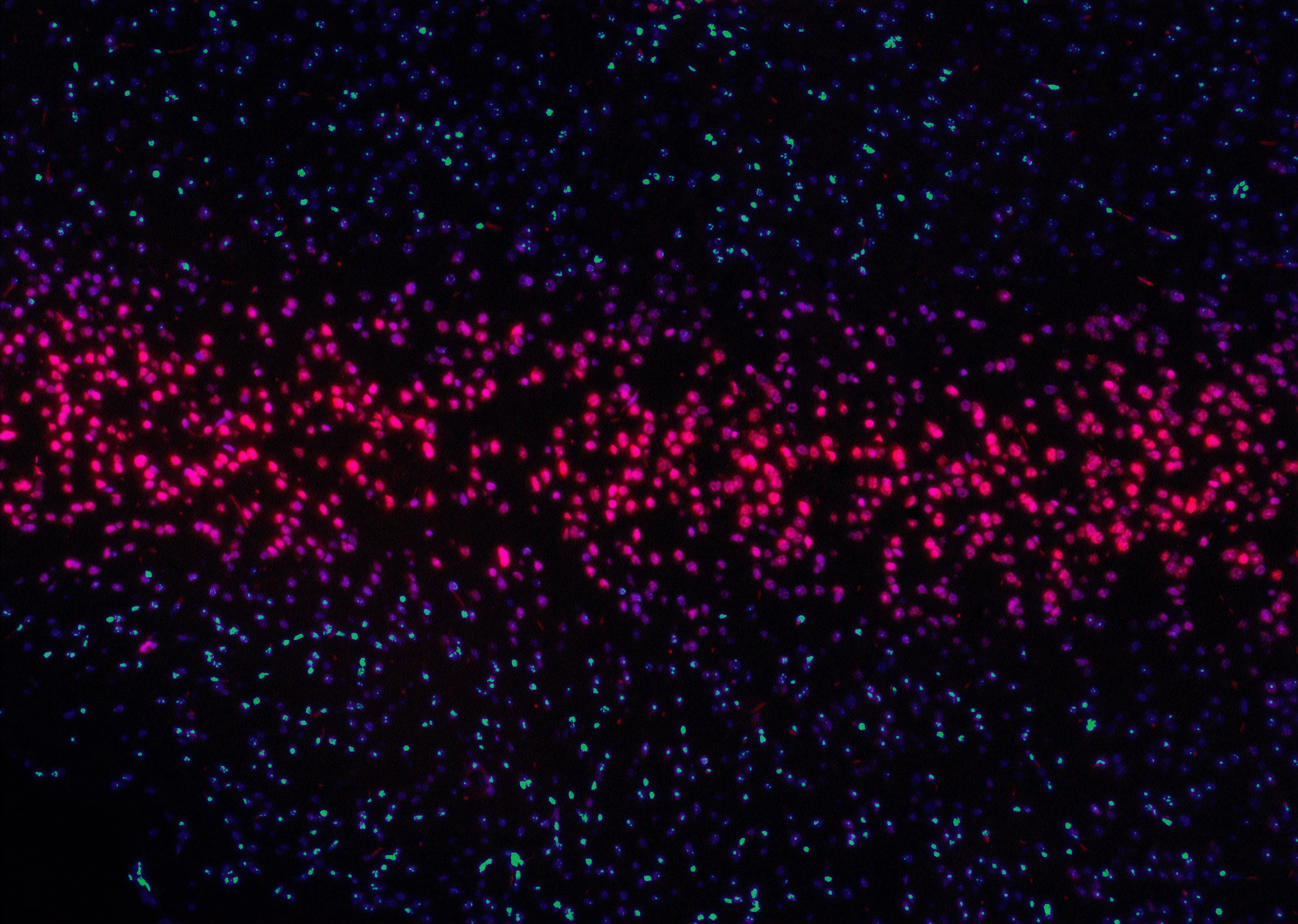
The research provides evidence of the conditions that trigger glial cells to consume synapses and promote neuroinflammation. To uncover the trigger, the postmortem brain tissue of 40 individuals who donated their brains to the Massachusetts Alzheimer’s Disease Research Center Brain Bank was examined. The samples were from three groups:
- The first group was cognitively normal without AD pathology (control group).
- The second group had a moderate amount of tau tangle pathology but no AD symptoms (resilient group).
- The third group had a moderate amount of tau tangle pathology and did display symptoms of dementia (AD group).
Both the second and third groups had a comparable amount of amyloid plague bundles.
Importantly, the study cohort was free of known comorbidities that otherwise could have contributed to an individual’s cognitive symptoms.
In AD, while amyloid plaques are found broadly throughout the brain, tau tangles spread along more typical patterns starting from the hippocampus. While tau tangles are relatively large and form inside neurons, smaller soluble tau species (called oligomers) can exit the neurons where they originate. By assessing a region of the brain in which tau tangles had not yet spread to, the visual cortex, the study discovered that in symptomatic cases, unlike in resilient or control cases, tau oligomers accumulated in synapses prior to the appearance of tau tangles in neurons. Tau oligomers triggered an excessive neuroinflammatory response by the brain’s glial cells. As these cells disproportionately activated in their response, they engulfed and eliminated synapses riddled with oligomeric tau. The study found that in clinically symptomatic AD, there was twice the level of tau oligomers within synapses serving as an “eat me” signal to microglia and astrocytes.
Using expansion microscopy, the study showed that in symptomatic cases, microglia and astrocytes consumed more synapses with tau oligomers than they did in resilient and control samples. Symptomatic cases were found to have approximately 40% fewer synapses compared with the resilient and control samples.
In summary, the study suggests that compared with resilient and control cases, in symptomatic AD there is an abnormal abundance of tau oligomers in synapses, and glial cells preferentially eliminate these synapses. Synaptic loss preceding tau tangle development could be highly important, since therapeutics targeting tau pathology have been prioritized due to the correlation in time between the development of tau tangles and the emergence of clinical symptoms. Additional studies are needed to fully understand the extent at which tau oligomers are toxic in AD. Given the abundant evidence that neuroinflammation plays a critical role in AD, the hope is that understanding and leveraging the influencers of resilience will allow for therapies for the prevention of cognitive decline.
Published in JAMA Neurology
Teresa Gomez-Isla, M.D., Harvard Medical School/Massachusetts General Hospital
Karen E. Duff, Ph.D., University College London, England
Tau Oligomer–Containing Synapse Elimination by Microglia and Astrocytes in Alzheimer Disease